00.Variants calling Q&A
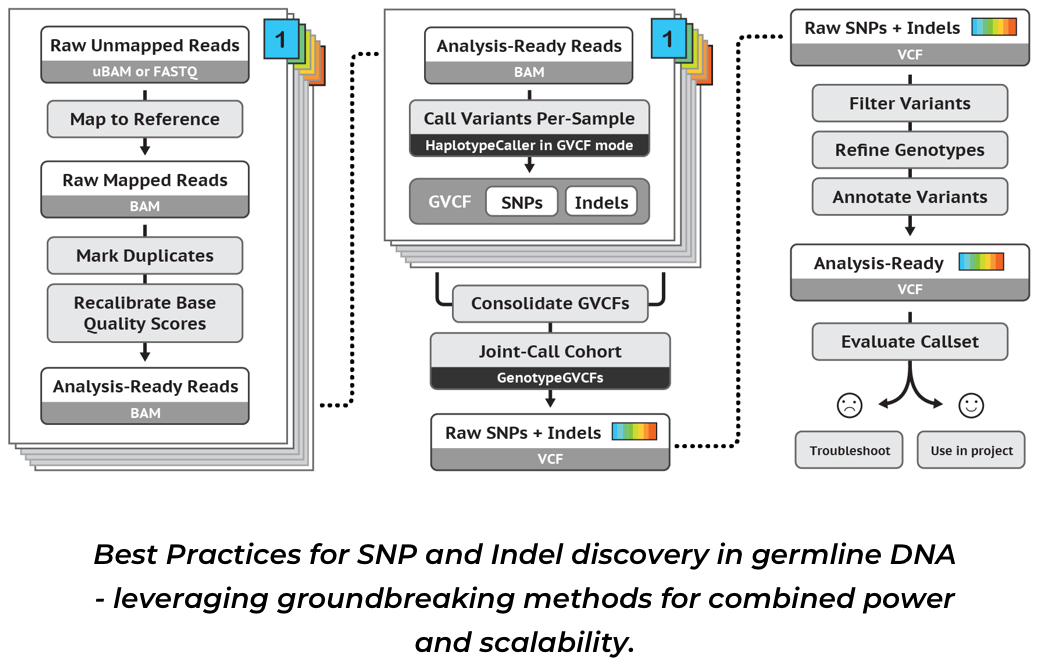
GATK best practice pipeline
为什么要去除/标记重复序列
需要去掉的是那些由于PCR扩增导致的重复序列,特点就是序列的起始和终止位点一致,这样的序列携带的信息是一致的,所以只保留一条即可。
BQSR
GATK的局部重比对
GATK会对indels热点区域进行局部的重新比对,这样可以避免由于indel导致的大量的假阳性。
但如果后续就是用HaplotypeCaller进行分析,这一步可以省去,因为HaplotypeCaller就是基于局部重比对。
GenomicsDBImport
GATK官方推荐的文件存储格式,据说性能更佳。
包含以下几个功能:
- Create a new GenomicsDB datastore from one or more GVCFs
- Joint-call samples in a GenomicsDB datastore
- Extract data from a GenomicsDB datastore
- Incrementally add new GVCFs to an existing GenomicsDB datastore
- Generate interval_list for an existing GenomicsDB datastore
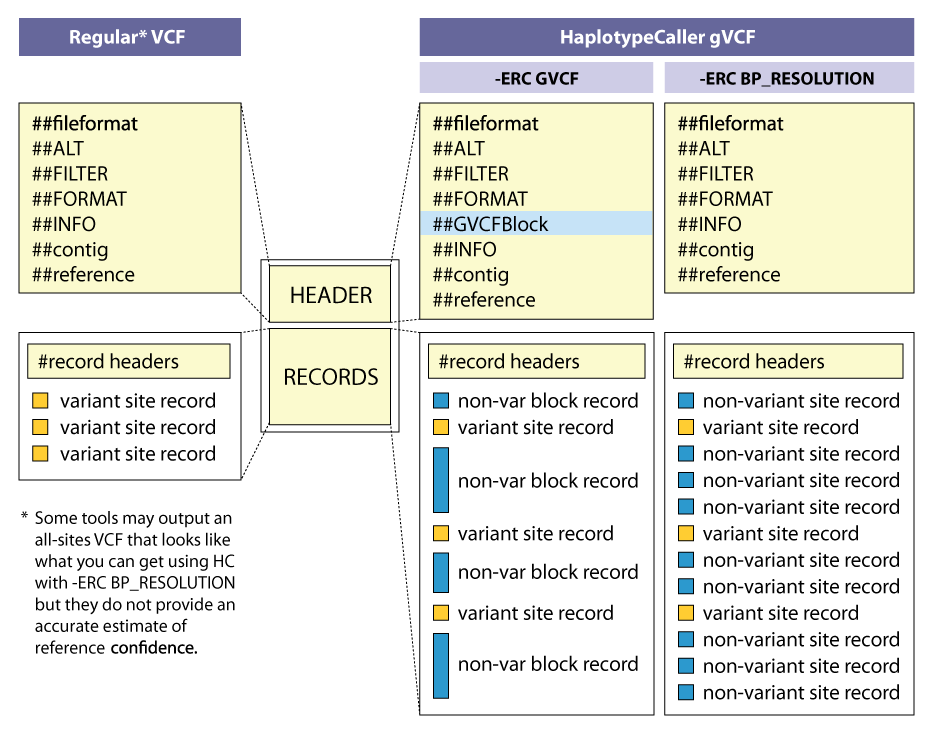
VCF和GVCF的区别
为什么采用Joint calling的方式
-
数据共享:在joint calling过程中,信息在所有样本间共享。这意味着如果一个样本在某个位点的测序覆盖度较低,其他样本中相同位点的高置信度变异可以辅助调用,从而提高低频变异的检测能力。
-
区分能力:joint calling能够更清晰地区分纯合参照位点和缺失数据位点。这是因为在joint calling中,只要调用集中的任何一个个体在某个位点有变异证据,就会在该位点输出基因型调用。
-
减少假阳性:joint calling通过使用统计模型对大量数据进行变异过滤,提高了过滤假阳性的能力。这种过滤通常比单独分析每个样本更有效,因为它可以在整个样本集上应用统一的过滤标准。
-
灵活性和扩展性:GATK 3.0及以上版本引入了增量joint calling的概念,即先对每个样本单独调用变异(生成GVCF文件),然后对所有样本的GVCF文件进行joint genotyping。这种方法解决了传统joint calling在计算资源和时间上的不足,同时保持了joint calling的优势。
-
处理大规模样本集:joint calling通过GenomicsDBImport工具和GenotypeGVCFs工具,可以高效地处理大规模样本集,使得对大型队列的研究成为可能。
-
解决N+1问题:在传统的joint calling中,每当有新的样本加入时,需要重新对所有样本进行分析。而增量joint calling允许对新样本进行单独分析,然后将其添加到现有的GVCF数据集中,无需重新分析所有旧样本。
VQSR
HardFilter
GATK针对单倍体怎么call SNP
--ploidy 1 \ # 关键参数:设置为单倍体
--heterozygosity 0.001 \ # 降低杂合度预期